Altered proteins in brain neurodegenerative diseases
In this new article series we aim at highlighting the current state of knowledge and the latest developments in the field of Alzheimer’s disease (AD) testing. This chapter looks at the main biomarkers used for IVD procedures.
Other articles in this series:
- Using CSF biomarkers to link pathology and clinical presentation
- How to perform a lumbar puncture
- Handling and transportation of CSF samples
- CSF biochemical pattern interpretation
- Aβ deposition and clearance: a key feature of ageing brain
- New criteria for Alzheimer’s disease
A biomarker is defined here as a characteristic that is objectively measured in body fluids with a level of evidence sufficient to be used in in vitro diagnostic procedures, i.e. evaluate disease risk, guide clinical diagnosis, monitor therapeutic interventions.
In neurodegenerative diseases, the biomarkers derived from the cerebrospinal fluid (CSF) has been intensely studied. The CSF biomarkers reflect molecular events in the brain due to it being in direct contact with the extracellular space of the brain.
Nevertheless, CSF movement is not a unidirectional flow and there is no evidence that the pathologic proteins can be found in the extracellular environment. The search of neurodegenerative disease (NDD) biomarkers is a challenge because most of brain proteins are extensively modified post-translationally.
Alzheimer-related biomarkers
Protein markers have been developed that reflect the central pathogenic processes in Alzheimer‘s pathology, i.e. the disturbance in the metabolism of β-amyloid (Aβ) and its subsequent deposition in senile plaques, the hyperphosphorylation of tau protein with subsequent formation of tangles (phosphorylated tau, P-tau) and the neuronal degeneration (total tau, T-tau).
Amyloid peptides
The main component of plaques is a peptide called β-amyloid (Aβ), which is a cleavage-metabolite of the amyloid precursor protein (APP). APP is a single-transmembrane protein with the Aβ domain partly embedded in the membrane. Aβ is generated by cleavage of APP by two proteases, the β- and γ-secretases. Free Aβ is secreted into the CSF.
There are two major C-terminal variants of Aβ, a shorter form ending at amino acid 40 (Aβ40) and a longer form ending at amino acid 42 (Aβ42). The Aβ42 isoform has a high tendency for aggregation, and is also the earliest Aβ species deposited into plaques.
Tau protein
Tau is a normal protein located in the neuronal axons in the brain. Its function is to stabilize the microtubular network in the axons, by binding to the microtubules. There are six different isoforms of tau, depending on which exons of the tau gene are translated to the mature tau protein. There are also numerous phosphorylation sites, i.e. amino acids that can be phosphorylated on the Tau protein.
The CSF level of T-tau reflects the intensity of the neuronal and axonal degeneration and damage in the brain.
Hyperphosphorylated Tau protein
In AD, a phosphate group is attached to several amino acids in tau protein, and tau is thus found in variants with different degrees of phosphorylation. Phosphorylated tau has a reduced ability to bind to the microtubules in the axons, which affect the axonal stability and thus the neuronal function, and also render tau an increased tendency for aggregation into paired helical filaments which then form the larger protein aggregates that make up the tangles.
Lewy-related biomarkers
Alpha-synuclein
Synucleinopathies are characterized by intra-neuronal aggregates consisting mainly of α-synuclein (α-syn) are found in Lewy bodies and Lewy neurites in Parkinson’s disease (PD), Parkinson’s disease dementia (PDD) and dementia with Lewy bodies (DLB) and in glial cytoplasmic inclusions in multiple system atrophy (MSA). This extracellular form of α-syn seems to be secreted from neuronal cells by exocytosis and detected in CSF as phosphorylated oligomeric α-syn.
Frontotemporal dementia associated biomarkers
The various FTD spectrum disorders are associated with brain accumulation of different proteins: tau, the transactive response DNA binding protein of 43 kDa (TDP43), or fused in sarcoma (FUS) protein, Ewing sarcoma protein and TATA-binding protein-associated factor 15 (TAF15) (the latter three are collectively known as FET proteins).
TAR DNA-binding protein of 43kDa (TDP-43)
TDP-43 is a major component of ubiquitin-positive inclusions that are one of the neuropathological hallmarks in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). Only 50% of FTLD patients have aggregates positive for TDP-43. Unfortunately, TDP-43 in CSF originates mainly from blood. Measurements of TDP-43 in CSF and blood are of minor importance as a diagnostic tool, but may be important for monitoring therapy effects of TDP-43 modifying drugs in the future.
FET/FUS proteinopathies
Recent reports identified mutations causative of neurological disorders in the genes encoding a family of RNA-binding proteins (RBPs) named FET. RNA-binding proteins (RBPs) are involved at all stages of RNA metabolism in neurodegenerative diseases. FET proteins are highly conserved and ubiquitously expressed. Recently, it has been suggested the involvement of FET proteins in neurological diseases, such as frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS), where they have been found in cytoplasmic aggregates. Abnormal co-accumulation of FET proteins into pathological inclusions has been described in all subtypes of FTLD-FUS. FUS (fused in sarcoma) is the most examined protein; FUS is localized in dendritic granules and spines in neurons where it plays a role in mRNA transport into dendrites, which represents an essential process for local protein synthesis and synaptic plasticity. No CSF FET related-markers are available.
Prion protein associated deposits
Pathological prion protein (PrPSc)
Human prion diseases are rapidly progressive neurodegenerative disorders caused by prion protein misfolding. The sporadic Creutzfeldt-Jakob disease (sCJD) is the most common form (85–90% of cases), followed by genetic CJD (gCJD) and fatal familial insomnia (FI) (10–15% of cases), which are linked to point or insertion mutations in the prion protein gene (PRNP). Several molecular subtypes of sporadic Creutzfeldt–Jakob disease have been identified and electroencephalogram and cerebrospinal fluid biomarkers have been reported to support clinical diagnosis but with variable utility according to subtype (updated WHO criteria for the diagnosis of CJD and related disorders, 2009). The principle for detecting PrPSc is to exploit the ability of small amounts of CSF PrPSc to convert native PrP into PrPSc in a newly described protein aggregation assay known as real-time quaking-induced conversion (RT-QuIC). This technique using a recombinant PrP showed good diagnostic sensitivity (82-96%) and virtually full specificity.
Prion diseases may trigger biochemical changes similar to AD involving PrPSc, Aβ42, APOE-4 and abnormal tau. Autopsied brain of sCJD showed also Alzheimer disease (AD)-like changes (17% of cases).
Testing of 14-3-3 protein in CSF is a standard biomarker test in suspected sCJD diagnosis by established Western blot method in CJD reference laboratories. Blood-contaminated samples which may result in artificially elevated CSF levels of 14-3-3.
Additional biomarkers of any cause of neuronal damage or injury
Neurofilaments as marker of axonal injury
Neurofilaments are intracellular intermediate filaments found in the central and peripheral nervous systems. In neurons, they control axonal diameter, which is correlated with nerve conduction velocity. Neurofilament protein include three subunits: neurofilament light (NfL) chain of ~68 kDa, Nf medium chain of ~150 kDa, and Nf heavy chain of ~190– 210 kDa. After axonal injury, intracellular neurofilaments can leak into the extracellular space, leading to an increased concentration in the CSF. Comparable performance of NfL in blood and CSF demonstrates its promise as a noninvasive biomarker of neurodegeneration.
Astroglial biomarker
S100B protein is an astroglial 11 kDa calcium-binding protein. In the classic neurodegenerative disorders (AD, PD, ALS) S100B concentration in CSF usually reflects the severity of the pathological condition, whereas, in many cases, S100B levels in blood remains unchanged during the course of the disease. However, serum S100B is valuable in the assessment of mild head injuries.
Neuronal damage biomarker
Neuron specific enolase (NSE) is a glycolytic isoenzyme located in central and peripheral neurons and neuroendocrine cells. The measurement of NSE in CSF could be a sensitive index of neuronal damage. NSE is considered a biomarker of neuronal stress and has prognostic potential for a variety of neurological disorders. Serum NSE levels are significantly elevated in patients with unfavorable neurological outcome in a variety of conditions.
Inflammation in neurodegeneration
Triggering receptor expressed on myeloid cells 2 (TREM2) is a transmembrane protein that is specifically expressed on microglia in the brain. TREM2 is one of the most crucial factors in regulating the innate immune system during AD progression. Soluble TREM2 (sTREM2) is the ectodomain released in a soluble form. sTREM2 is described to be a central regulator of microglial function and CSF sTREM2 is known to increase 5 years before the expected symptom onset in AD.
Bibliography
- TREM2 ectodomain and its soluble form in Alzheimer’s disease. Yang J, et al. J Neuroinflammation. 2020; 17(1): 204. Review.
- New Insights into the Role of Neuron-Specific Enolase in Neuroinflammation, Neurodegeneration, and Neuroprotection. Haque A, et al. Brain Sci. 2018; 8(2): 33. Review.
- The S100B story: from biomarker to active factor in neural injury. Michetti F, et al. J Neurochem. 2019; 148(2): 168-187. Review.
- Scandinavian guidelines for initial management of minimal, mild and moderate head injuries in adults: an evidence and consensus-based update. Undén J, et al. Scandinavian Neurotrauma Committee (SNC). BMC Med. 2013; 11: 50. Practice Guideline.
- The diagnostic performance of neurofilament light chain in CSF and blood for Alzheimer’s disease, frontotemporal dementia, and amyotrophic lateral sclerosis: A systematic review and meta-analysis.Forgrave LM, et al. Alzheimers Dement (Amst). 2019; 11: 730-743.
- A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers.Jack CR Jr, et al. Neurology. 2016; 87(5): 539-547. Review.
- Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine. Kovacs GG. Int J Mol Sci. 2016; 17(2): 189. Review.
- Longitudinal CSF biomarkers in patients with early Parkinson disease and healthy controls. Mollenhauer B, et al. Parkinson’s Progression Marker Initiative. Neurology. 2017; 89(19): 1959-1969.
- Post mortem cerebrospinal fluid β-synuclein levels are raised in multiple system atrophy and distinguish this from the other β-synucleinopathies, Parkinson’s disease and Dementia with Lewy bodies. Foulds PG, et al. Neurobiol Dis. 2012; 45(1): 188-195.
- Neurochemical biomarkers in the diagnosis of frontotemporal lobar degeneration: an update. Oeckl P, et al. J Neurochem. 2016; 138 Suppl 1: 184-192. Review.
- Limited role of free TDP-43 as a diagnostic tool in neurodegenerative diseases. Feneberg E, et al. Amyotroph Lateral Scler Frontotemporal Degener. 2014; 15(5-6): 351-356.
- Role of FET proteins in neurodegenerative disorders. Svetoni F, et al. RNA Biol. 2016; 13(11): 1089-1102. Review.
- Prion specific and surrogate CSF biomarkers in Creutzfeldt Jakob disease: diagnostic accuracy in relation to molecular subtypes and analysis of
neuropathological correlates of p tau and Aβ42 levels. Lattanzio F, et al. Acta Neuropathol. 2017; 133(4): 559–578. - Cerebrospinal fluid real-time quaking-induced conversion is a robust and reliable test for sporadic creutzfeldt-jakob disease: An international study. McGuire LI, et al. Ann Neurol. 2016; 80(1): 160-165. Multicenter study.
- Validation of 14-3-3 Protein as a Marker in Sporadic Creutzfeldt-Jakob Disease Diagnostic. Schmitz M, et al. Mol Neurobiol. 2016; 53(4): 2189-2199.
- Progress in CSF biomarker discovery in sCJD. Llorens F, et al. Oncotarget. 2017; 8(4): 5666-5667. Editorial.
- Cerebrospinal β-synuclein in β-synuclein aggregation disorders: tau/αsynuclein ratio as potential biomarker for dementia with Lewy bodies. Llorens F, et al. J Neurol. 2016; 263(11): 2271-2277.
- Prion Disease Induces Alzheimer Disease-Like Neuropathologic Changes. Tousseyn T, et al. J Neuropathol Exp Neurol. 2015; 74(9): 873-888.
Related articles

Integration of ADx NeuroSciences within Fujirebio unleashes a world of new possibilities
Fujirebio Europe N.V. will formally and fully integrate ADx NeuroSciences N.V. into its organization on April 1, 2026. This completes a process that...

Fujirebio introduces its Neuro Expert Toolbox (NExT) at AAIC 2025
Fujirebio is introducing its Neuro Expert Toolbox (NExT) for the first time at AAIC 2025 (Alzheimer's Association International Conference®) in...

Video - Alzheimer's awareness redefined
Follow the journey of the Sullivan family and leading Alzheimer’s Neurologist and Researcher Dr. David Greeley as they introduce and explain these...

Publication - Serum and cerebrospinal fluid neurofilament light chains measured by SIMOA™, Ella™, and Lumipulse™ in multiple sclerosis naïve patients
We would like to draw your attention to a first publication on our Lumipulse® G NfL solution. In this article you will find a method comparison of CSF...

CTAD 2023 – Spotlight on recent advances in blood-based biomarkers for Alzheimer’s disease
Both fluid and imaging biomarkers provide biological evidence for the underlying etiology of cognitive impairment. The core fluid biomarkers of...

IVDR status update for Fujirebio’s Neuro products
The European CE-marking is used to support registrations of in vitro diagnostic (IVD) medical devices in many jurisdictions around the world. The...
Scientific poster - Blood sample matrix validation, impact of sample freezing and method comparison analysis using the Lumipulse® G NfL blood prototype assay
This AD/PD 2023 poster investigates the agreement between matched serum and plasma samples on the Lumipulse G NfL Blood prototype assay, the impact of...

Video - A neurochemist's search to save memories
Meet Dr. Charlotte Teunissen, Professor in Neurochemistry, and her lifelong friend Christa Reinhoudt, who was diagnosed with Alzheimer's disease in...
Scientific poster - Analytical performance of the Lumipulse® G NfL CSF <RUO>
Poster presented at the AD/PD 2023
This AD/PD 2023 poster wishes to demonstrate the analytical performance of the newly developed Lumipulse G NfL CSF...
Scientific poster - CSF pTau181/Aβ1-42 ratio increases pre-analytical variability over measuring Aβ1-42 alone
In this CTAD 2022 poster, we examine the utility of CSF biomarker ratios to correct for pre-analytical variability.
Scientific poster - A fully automated and scalable plasma pTau181 assay for Alzheimer's disease
In this article, the diagnostic performance of a modified version of the Lumipulse G pTau181 CSF test is evaluated.

New criteria for Alzheimer’s disease
New criteria for different stages of AD have been suggested by the International Working Group (IWG) and the National Institute on Aging-Alzheimer’s...
Aβ deposition and clearance: a key feature of the ageing brain
This chapter looks closer at Aβ deposition and clearance as key feature of ageing brain.
Scientific poster - Reducing misdiagnosis of Alzheimer’s disease pathology utilizing CSF and amyloid PET
In this poster we examine the performance of cognitive testing alone for identification of amyloid positivity in MCI patients from the ADNI study when...
Scientific poster - Analytical performance overview of the Lumipulse® G pTau 181 Plasma RUO assay
The aim of the study, presented at the AAIC 2022, was to determine the performance of several analytical parameters, including amongst others...
Scientific poster - Analytical performance of the Lumipulse® G β-Amyloid 1-40 Plasma and Lumipulse® G β-Amyloid 1-42 Plasma RUO assays
The aim of the study, presented at the AAIC 2022, was to determine the performance of several analytical parameters, including amongst others...
CSF biochemical pattern interpretation
What are some of the best-practices of CSF biochemical pattern interpretation? In this article series we aim at highlighting the current state of...
Handling and transportation of CSF samples
Cerebrospinal fluid (CSF) can be collected in the lumbar region by an experienced physician. This article details the recommended procedure for...

How to perform a lumbar puncture
In this article series we aim at highlighting the current state of knowledge and the latest developments in the field of Alzheimer’s disease (AD)...
Using CSF biomarkers to link pathology and clinical presentation
In this article series we aim at highlighting the current state of knowledge and the latest developments in the field of Alzheimer’s disease (AD)...

Video - A day at the Fujirebio Neuro Center of Excellence
In this short video we show you around at the Fujirebio Neuro Center of Excellence. Right now, expectations are high for the development of blood...
Scientific poster - Comparing CSF and plasma LUMIPULSE® Alzheimer’s Disease biomarker analysis to amyloid-β PET imaging
The aim of this study was to evaluate a plasma pTau biomarker as a tool for predicting amyloid pathology.
Improving clinical diagnosis of Alzheimer’s disease: Review of the available literature
In this chapter, we will review available literature on the accuracy of the underlying pathological determinations in mild cognitive impairment (MCI)...
The drawbacks of relying solely on the standard routine clinical examinations and cognitive testing
Many subtypes of disease exist under the umbrella of dementia with Alzheimer’s disease (AD) being the most common. AD-related dementia is...

Testimonial - Value of the β-Amyloid ratio and other CSF biomarkers in the Erlangen Score interpretation algorithm
By Prof. Dr. Piotr Lewczuk - Two groups of established cerebrospinal fluid (CSF) biomarkers reflect two major pathological alterations in Alzheimer's...
Scientific poster - Towards an easy plasma pTau 181 detection
Blood-based Alzheimer’s disease (AD) biomarker testing could be used as a simple, accessible, and scalable approach to help support the diagnosis of...
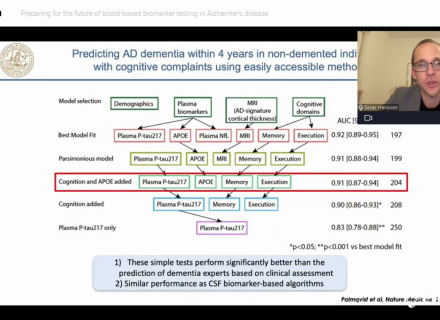
Webinar replay - Preparing for the future of plasma based Alzheimers disease diagnostics
At Fujirebio we are hosting a webinar series with leading expert speakers dedicated to current topics in the field of Alzheimer's disease diagnostics...
Video - The interest of automated testing for all four CSF biomarkers
In less than 2 minutes, this video explains the advantages of automated biomarker testing for all four CSF biomarkers, over other available testing...

The Fujirebio Neuro Center of Excellence
The Fujirebio Neuro Center of Excellence has been founded with the objective of rising to this challenge. It is a research and development hub focused...
Booklet - First edition of our new clinical booklet "A few drops of insight can lead to an ocean of understanding"
Early diagnosis of Alzheimer's disease is crucial. The desire to tackle neurodegenerative diseases by always finding earlier diagnostic solutions and...
Video - Ratio calculation of Aβ1-42 and Aβ1-40 offers essential information about the buildup of amyloid pathology in a patient's brain
This 2 minute video explains why and how a ratio calculation of the two amyloid proteins, Aβ1-42 and Aβ1-40, offers particularly essential information...

Video - Daniel's story about his early testing and diagnosis of Alzheimers disease
Daniel lives in Stockholm, Sweden, and was diagnosed with Alzheimer’s disease when he was still in his early 50's. In this 6-minute video we follow...

Influence of automation on Aβ1-42/Aβ1-40 ratio and its use
Automation is an important step in the direction of more standardization as it limits the number of manual handling steps and therefore minimizes...

Comparison of Aβ1-42/Aβ1-40 ratio with other ratios
CSF Aβ1-42/Aβ1-40 is a tool to normalize values of patients with different amyloid levels, as other ratios might be seen more as interpretation tools...
How to work with Aβ1-42/Aβ1-40 ratio
One cause of discordant results can be the preanalytical conditions, e.g. when laboratories use tubes that bind certain proteins. Aβ1-42 adsorption is...

Improvement of AD risk scores by use of the Aβ1-42/Aβ1-40 ratio
Different scores have been developed to provide an interpretation of biomarker results for AD diagnosis or risk prediction. Here we will give two...

Aβ1-42/Aβ1-40 ratio for interpretation of discordant results
By use of the Aβ1-42/Aβ1-40 ratio, discordant results (i.e. when amyloid and tau biomarkers are not concordant) can be improved. However, while the...

Use of the Aβ1-42/Aβ1-40 ratio to improve accuracy of AD diagnosis
It is widely agreed that, since cerebrospinal fluid (CSF) is in direct contact with the central nervous system (CNS), analytes measured in this body...

What is Alzheimer's disease?
Alzheimer’s disease, which is the most common form of dementia, is an incurable degenerative disease. Neurons in certain parts of the brain are...

 Powered by Bioz
Powered by Bioz















 and Lumipulse G pTau 181 Calibrators set (230367)")






 and Lumipulse G β-Amyloid 1-40 Calibrators (231531)")

 and Lumipulse G β-Amyloid 1-42 Calibrators (230343)")