KL-6 in Lung Diseases: Current classification of interstitial lung diseases
By Dr. Sandra Langer, Fujirebio
Welcome to a new series of Insight posts about KL-6 in Lung Diseases!
Interstitial lung diseases (ILDs) represent a large, heterogeneous group of more than 200 different entities, most of which are classified as rare diseases.
Accurate diagnosis of Idiopathic Pulmonary Fibrosis (IPF) is critical, as other forms of ILD that have similar clinical presentations to IPF require different treatment strategies.
Imaging plays an essential role in the diagnosis of ILDs.
Several scientific authors deal with the idea of combining IPF with other forms of fibrosing ILD that have, i.e. self-sustaining fibrosis, progressive decline in lung function, and early mortality in the group of “progressive fibrosing ILDs” that would describe ILD in patients who, independent of the classification of the ILD, at some point in time exhibit a progressive fibrosing phenotype.
Pulmonary function parameters at a single time point do not reliably predict disease behavior and, despite multiple attempts, High resolution computer tomography (HRCT)-quantified disease extent on sequential imaging has not been established as a reliable marker of disease progression.
Based on the results from a number of reports investigating Krebs von den Lungen-6 / Mucin 1 (KL-6/MUC1), the serum levels of KL-6/MUC1 are useful for 1) detecting the presence of disease, 2) evaluating disease activity, and 3) predicting outcomes in various types of ILDs.
Read the first post below or download the complete guidance document now!
You can follow our Insight articles over the next weeks and get some deeper information on the value of measuring KL-6 in the field of lung diseases. But you can also directly download the complete guidance document right away, following this link (requires a Premium eServices account).
We are always happy to hear comments and questions on this topic, so feel free to contact us. Many thanks!
- You will find more resources about ILDs and the KL-6 biomarker in our dedicated microsite at www.fujirebio.com/kl-6 (opens in a new window).
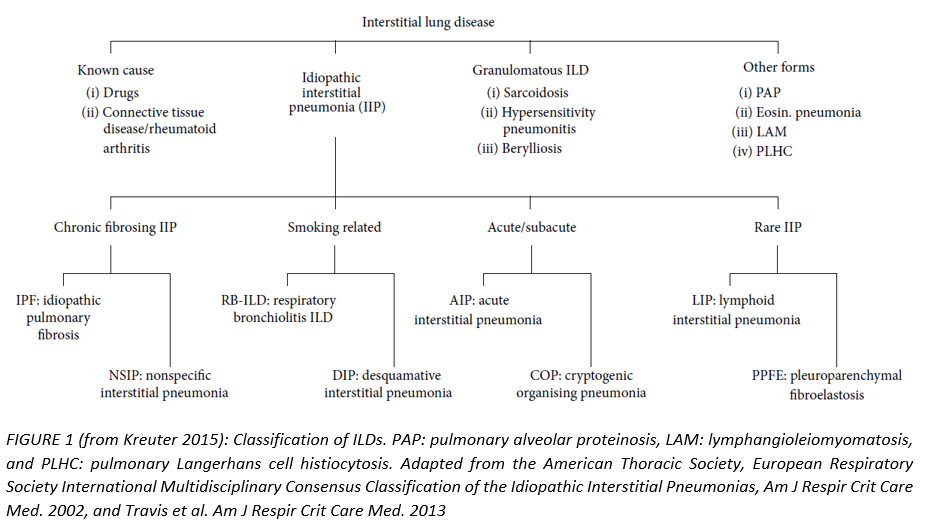
Current Classification of interstitial lung diseases
- ILD early and accurate diagnosis is challenging due to heterogeneity of the disease.
- Even with multidisciplinary team assessment, 15-25% of ILD patients remain unclassifiable.
Interstitial lung diseases (ILDs) represent a large, heterogeneous group of more than 200 different entities, most of which are classified as rare diseases. They are defined as lung diseases that affect the alveolar structures, the pulmonary interstitium, and small airways. The current diagnosis of an ILD relies mainly on the combination of clinical, radiological, and pathological criteria, which should be explored in a multidisciplinary board.2
Even if Interstitial Pulmonary Fibrosis (IPF) can be seen as the largest group within ILDs it has been shown that approximately one in 10 patients has an unclassifiable ILD. This makes it the fourth most common classification behind IPF (21%), hypersensitivity pneumonitis (15%) and sarcoidosis (14%).3
While multidisciplinary team assessment yields a definite diagnosis in many cases of interstitial lung disease, 15-25% of patients remain unclassifiable.1
References
- Bendstrup, Elisabeth, et al. "Challenges in the classification of fibrotic ILD." Sarcoidosis vasculitis and diffuse lung disease 32.1S (2015): 4-9.
- Kokosi, Maria A., George A. Margaritopoulos, and Athol U. Wells. "Personalised medicine in interstitial lung diseases: Number 6 in the Series “Personalised medicine in respiratory diseases” Edited by Renaud Louis and Nicolas Roche." European Respiratory Review 27.148 (2018): 170117.
- Ryerson, Christopher J., et al. "Prevalence and prognosis of unclassifiable interstitial lung disease." European Respiratory Journal 42.3 (2013): 750-757.
Don't miss the next posts on this topic in the following weeks:
- ILD diagnosis – current professional practice
- Use of serum biomarkers in progressive fibrosing ILDs
- KL-6 in disease prognosis
- From Classification to disease activity
You might also find this website interesting:
- You will find more resources about ILDs and the KL-6 biomarker in our dedicated microsite at www.fujirebio.com/kl-6 (opens in a new window).
Related articles
[Jan 2022 Update] This scientific literature overview demonstrates the excellent performance of the fully automated nucleocapsid protein antigen assay Lumipulse® G SARS-CoV-2 Ag
Lumipulse G SARS-CoV-2 Ag was the first high-sensitive nucleocapsid protein antigen assay launched on a fully automated chemiluminescent platform.
...

Webinar: Lumipulse® G KL-6, the biomarker for routinely assessment of epithelial alveolar damage in pulmonary fibrosis and COVID-19
During this webinar Dr. Francesco Bonella (MD - Center for Interstitial & Rare Lung Disease, Department of Pulmonology, Ruhrlandklinik University...

Japanese authorities have chosen Lumipulse® for nasopharyngeal or saliva-based SARS-CoV-2 antigen testing of passengers at their main international airports
Several fully automated and CLEIA-based LUMIPULSE G1200 systems from Fujirebio are right now performing fast, high sensitivity SARS CoV-2 antigen...
Can KL-6 be a specific marker of alveolar damage for COVID-19 patients?
There are many reference centers across Europe that have included KL-6 as one of the biomarkers within the COVID-19 protocol to strengthen the battery...

From Classification to disease activity
While IPF is the classic fibrosing ILD, clinical data suggest that there is a larger group of patients with differing clinical ILD diagnoses who...

KL-6 in disease prognosis
Recent research has shown that serum and bronchoalveolar lavage fluid level of KL-6 has an important value in the diagnosis, treatment assessment and...
Use of serum biomarkers in progressive fibrosing ILDs
Based on the results from a number of reports investigating KL-6/MUC1, the serum levels of KL-6/MUC1 are useful for 1) detecting the presence of...

ILD diagnosis – current professional practice
Accurate diagnosis of IPF is critical, as other forms of ILD that have similar clinical presentations to IPF require different treatment strategies.

Mesothelin, the novel, non-invasive in vitro biomarker to aid in the diagnosis of Mesothelioma
Mesothelioma is a rare form of cancer that is linked to exposure to asbestos. Although malignant mesothelioma remains a relatively uncommon malignancy...





 Powered by Bioz
Powered by Bioz")


